
氧化锂(Li2O)因其高锂含量、优异的化学稳定性与简单的反萤石结构,在固态离子导体、核聚变氚增殖材料及高温功能陶瓷等领域受到广泛关注。作为典型的超离子导体模型体系,Li2O在高温下发生亚晶格融化的二阶相变,呈现独特的离子动力学特征。尽管早期实验和模拟研究揭示了其在高温下的超离子行为,但离子扩散机制、超离子相变与缺陷动力学之间关系仍缺乏系统理解,构效关系尚不明确。传统第一性原理分子动力学受限于时间与尺度,而经验势模型精度不足,难以对宽温度区间的行为进行精确刻画。这一矛盾使得对Li2O的高温动力学过程进行精确刻画仍具挑战性。
为揭示Li2O在不同温度区间的扩散行为与相变机制,研究团队采用基于第一性原理训练的深度势能(Deep Potential,DP)模型结合分子动力学模拟,在保持第一性原理精度的同时实现纳秒级动力学分析,该高精度DP模型能够在宽温度范围内准确再现Li2O的结构与能量特征,为深入研究其动力学过程提供了可靠基础。 基于该模型的分子动力学模拟揭示了Li2O在升温过程中经历的三个阶段:常规固态、超离子态和液态。研究发现,在约1300 K附近,Arrhenius曲线出现明显弯曲,活化能发生变化,标志着Li2O进入超离子态;当温度升至1700 K以上时,扩散系数发生突变,Li2O发生一阶相变进入液态。
为了深入理解这一过程,团队深入分析了Li2O的弹性常数随温度的变化、扩散动力学轨迹和van Hove自关联函数。结果表明,在1100 K附近出现的C11弹性软化反映了离子运动的显著加速,而1300 K附近的超离子转变则源于Li亚晶格的熔化与集体扩散模式的形成。进一步的统计分析显示,高温下Li2O的超离子行为主要源自于[LiO4]四面体Li(LiTet)迁移至[LiO6]八面体(LiOct),产生Frenkel缺陷对。其中 LiOct展现出更高的扩散活性,在诱导超离子相变中起主导作用。
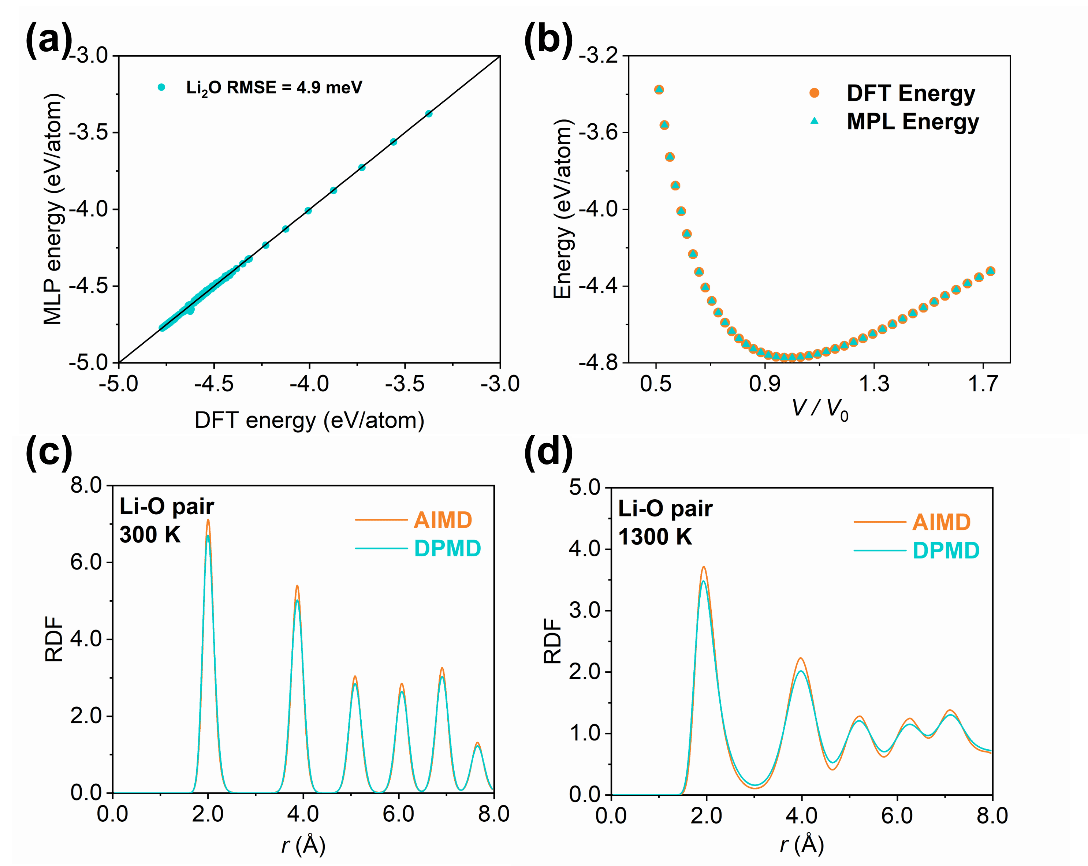
图1. DP势函数准确性。(a)DFT与DP能量对比。(b)能量-体积关系,DFT与DP 对比。(c)300 K下Li-O RDF,AIMD与DP对比。(d)1500 K下Li-O RDF,AIMD与DP对比。
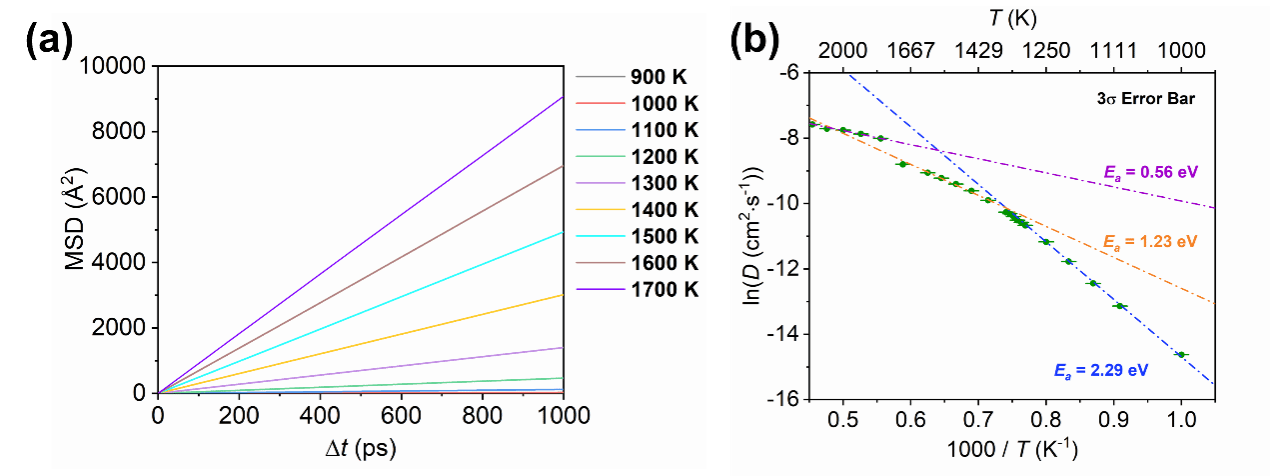
图2. Li2O扩散行为。(a)时间平均均方根位移(t-MSD)。(b)Li2O中Li+自扩散的阿伦尼乌斯图(不同颜色直线表示不同区域的拟合结果)。
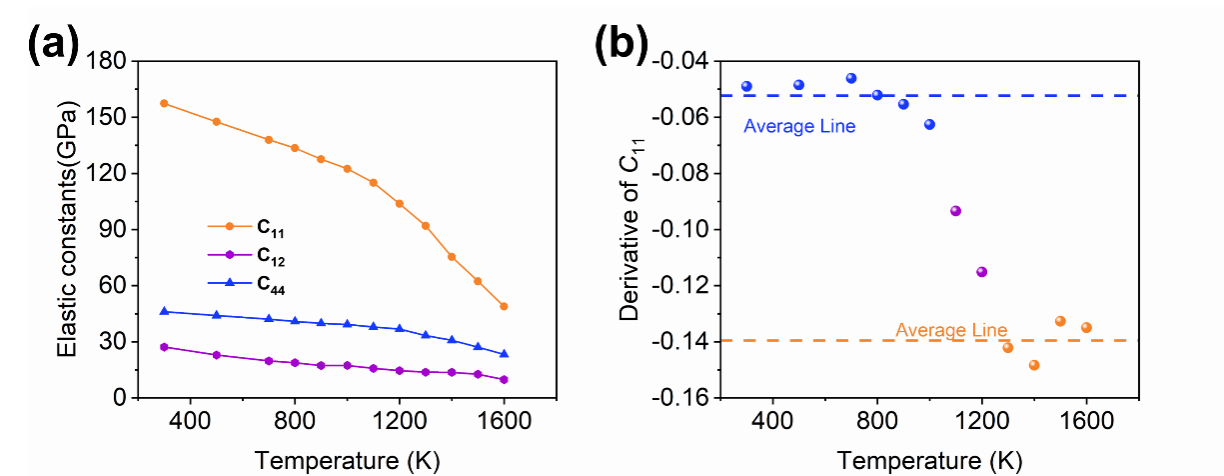
图3. Li2O弹性常数。(a)弹性常数随温度变化关系。(b)C11曲线斜率与温度关系。
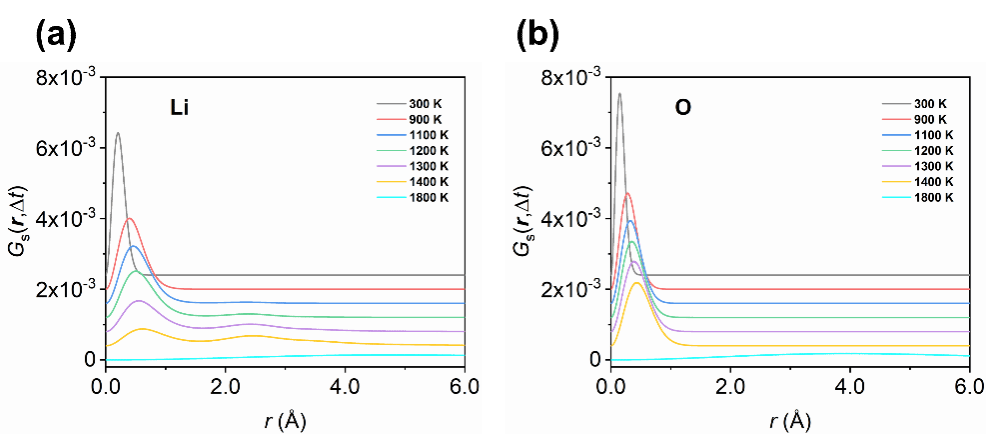
图4. 锂和氧的van Hove自关联函数。(a)锂的van Hove自关联函数。(b)氧的van Hove自关联函数。∆t设置为2 ps,所有曲线均进行了垂直平移以提高清晰度和便于比较。
研究团队在机器学习势函数的开发方面具有深厚的研究基础,基于主成分分析与主动学习结合的训练范式,已成功构建高精度的LLZO势函数,并将范式推广至其他体系。研究团队在机器学习势函数相关工作已发表十余篇论文。该论文所使用的势函数以该LLZO势函数为基础,进一步拓展引入Li2O相关结构数据集,通过多轮迭代训练完善模型的描述能力,显著拓展了其在复杂锂基体系中的适用范围。
该研究工作以 “Elastic softening and defect-mediated diffusion in superionic Li2O revealed by molecular dynamics” 为题发表在Physical Review B上(DOI: 10.1103/4z6n-zfdr)。物理学系2023级硕士生吴智丰为论文第一作者,吴顺情教授为通讯作者。厦门大学物理学系博士生游逸玮、硕士生张芳芳,及吕铁羽副教授、曹昕睿副教授、孙阳教授和朱梓忠教授等参与了相关研究工作。
文章链接: https://journals.aps.org/prb/abstract/10.1103/4z6n-zfdr




